Visualization tutorial¶
This tutorial show the visualization process of our processed Stereo-seq mosue midbrain dataset
The command for running on this dataset is:
ONTraC \
--meta-input data/stereo_seq_brain/original_data.csv \
--NN-dir output/stereo_seq_NN \
--GNN-dir output/stereo_seq_GNN \
--NT-dir output/stereo_seq_NT \
--device cuda \
-s 42 \
--lr 0.03 \
--hidden-feats 4 \
-k 6 \
--modularity-loss-weight 0.3 \
--regularization-loss-weight 0.1 \
--purity-loss-weight 300 \
--beta 0.03 2>&1 | tee log/stereo_seq.log
Note
The input dataset and output files could be downloaded from our Zenodo repository.
Preparation¶
Install Required Packages¶
pip install "ONTraC[analysis]"
Load Dataset¶
import requests
# URL of the file
url = "https://zenodo.org/records/15571644/files/Stereo_seq_data.zip"
# Local file path to save the file
file_path = "./Stereo_seq_data.zip"
try:
# Send a GET request to the URL
response = requests.get(url)
response.raise_for_status() # Check if the request was successful
# Write the content to the file
with open(file_path, "wb") as file:
file.write(response.content)
print(f"File downloaded and saved to {file_path}")
except requests.exceptions.RequestException as e:
print(f"An error occurred: {e}")
File downloaded and saved to ./Stereo_seq_data.zip
import zipfile
# Path to the zip file
zip_file_path = "Stereo_seq_data.zip"
# Directory where files will be extracted
extract_to_path = "./"
try:
# Open the zip file
with zipfile.ZipFile(zip_file_path, 'r') as zip_ref:
# Extract all files to the specified directory
zip_ref.extractall(extract_to_path)
print(f"Files extracted to '{extract_to_path}'")
except zipfile.BadZipFile:
print("The file is not a valid zip file.")
Files extracted to './'
Visualization through Command Line¶
ONTraC_analysis \
-o analysis_output/stereo_seq \
-l Stereo_seq_data/ONTraC_run_log/stereo_midbrain_base.log \
--NN-dir Stereo_seq_data/ONTraC_output/stereo_midbrain_base_NN \
--GNN-dir Stereo_seq_data/ONTraC_output/stereo_midbrain_base_GNN \
--NT-dir Stereo_seq_data/ONTraC_output/stereo_midbrain_base_NT \
-r
Visualization Step-by-step¶
If you want to adjust the figures, here is the example codes for step-by-step analysis using Python. We recommand you using jupyter notebook or jupyter lab here.
Load Modules¶
import matplotlib as mpl
mpl.rcParams['pdf.fonttype'] = 42
mpl.rcParams['ps.fonttype'] = 42
mpl.rcParams['font.family'] = 'Arial'
import matplotlib.pyplot as plt
import seaborn as sns
from ONTraC.analysis.data import AnaData
Plotting Preparation¶
from optparse import Values
options = Values()
options.NN_dir = 'Stereo_seq_data/ONTraC_output/stereo_midbrain_base_NN'
options.GNN_dir = 'Stereo_seq_data/ONTraC_output/stereo_midbrain_base_GNN'
options.NT_dir = 'Stereo_seq_data/ONTraC_output/stereo_midbrain_base_NT'
options.log = 'Stereo_seq_data/ONTraC_run_log/stereo_midbrain_base.log'
options.reverse = True # Set it to False if you don't want reverse NT score
options.output = None # We save the output figure by our self here
ana_data = AnaData(options)
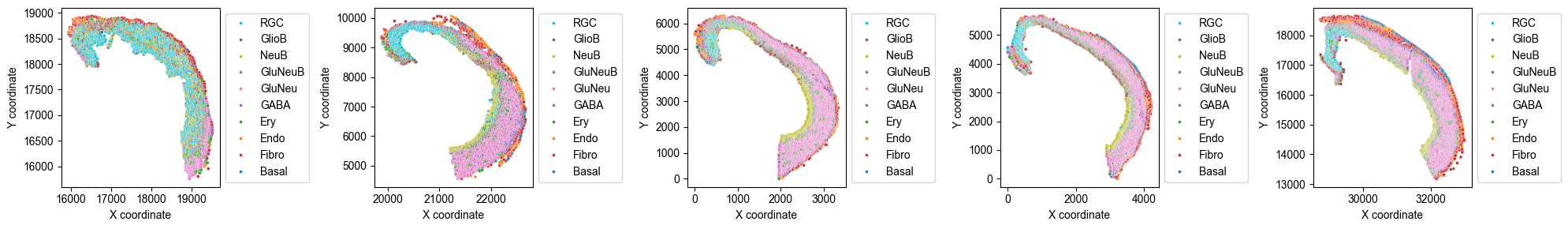
Spatial Cell Type Distribution¶
from ONTraC.analysis.cell_type import plot_spatial_cell_type_distribution_dataset_from_anadata
fig, axes = plot_spatial_cell_type_distribution_dataset_from_anadata(ana_data = ana_data,
hue_order = ['RGC', 'GlioB', 'NeuB', 'GluNeuB', 'GluNeu', 'GABA', 'Ery', 'Endo', 'Fibro', 'Basal'])
for ax in axes:
# ax.set_aspect('equal', 'box') # uncomment this line if you want set the x and y axis with same scaling
# ax.set_xticks([]) # uncomment this line if you don't want to show x coordinates
# ax.set_yticks([]) # uncomment this line if you don't want to show y coordinates
pass
fig.tight_layout()
# fig.savefig('figures/Spatial_cell_type.png', dpi=150) # uncomment this line if you want save the results
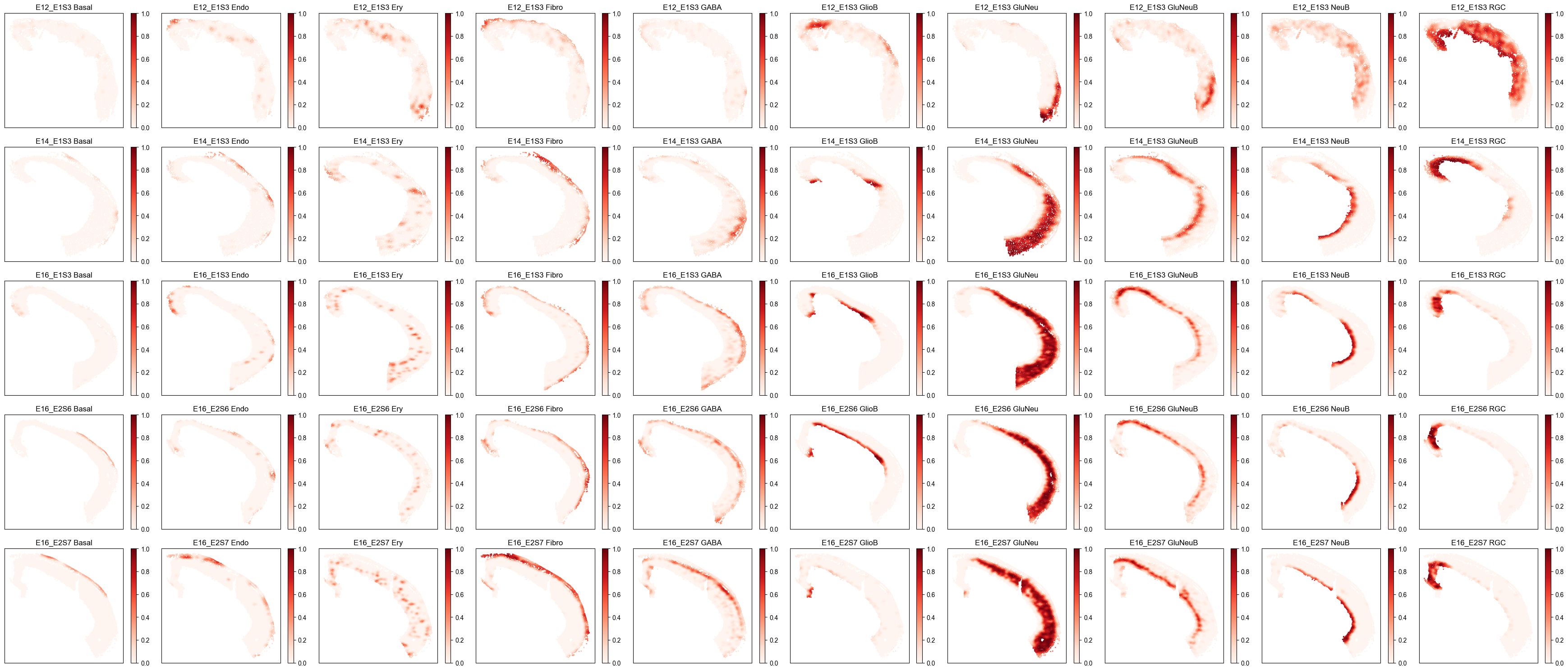
Spatial Cell Type Composition Distribution¶
from ONTraC.analysis.spatial import plot_cell_type_composition_dataset_from_anadata
fig, axes = plot_cell_type_composition_dataset_from_anadata(ana_data=ana_data)
# fig.savefig('figures/cell_type_compostion.png', dpi=100) # uncomment this line if you want save the results
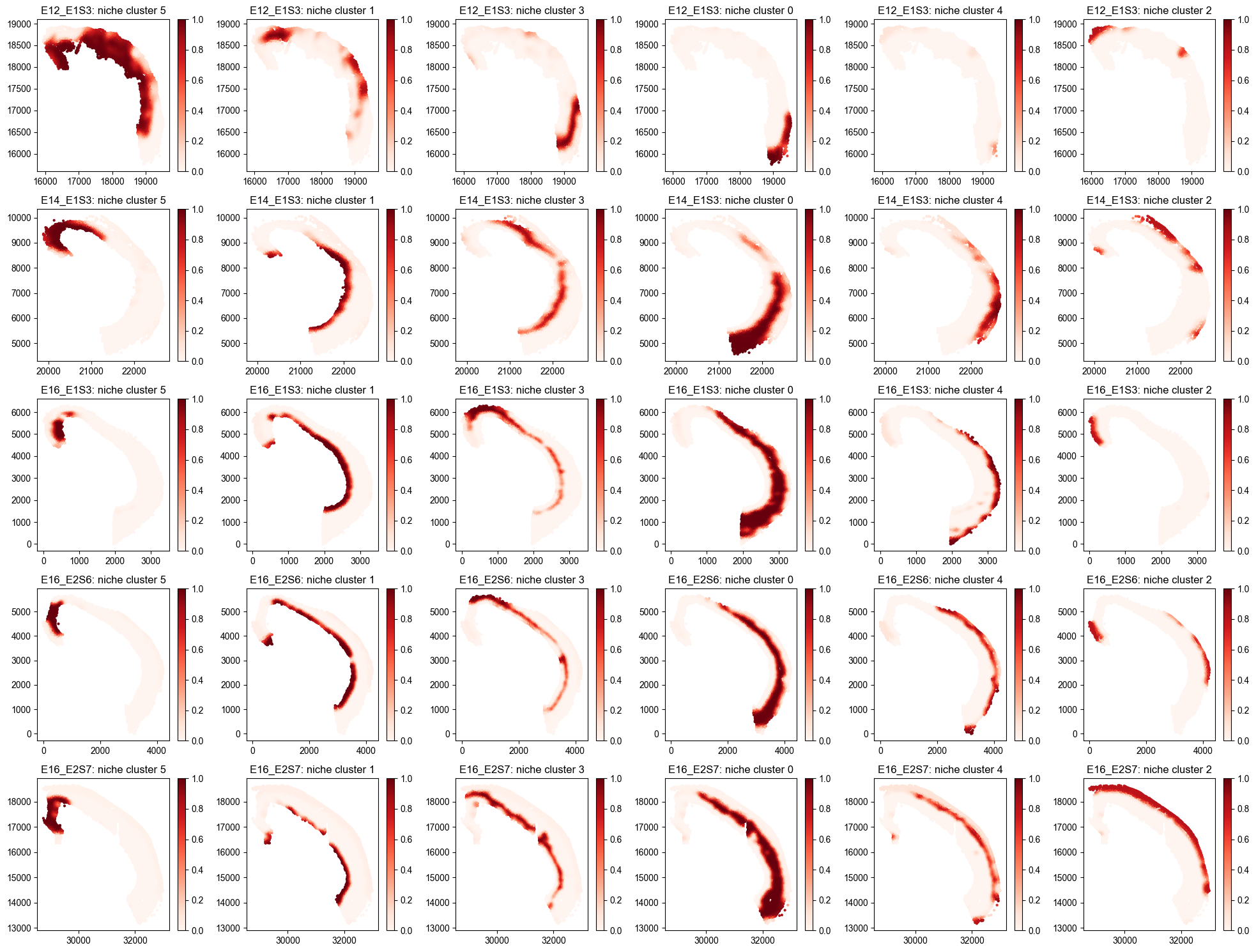
Niche Cluster¶
Spatial Niche Cluster Loadings Distribution¶
from ONTraC.analysis.niche_cluster import plot_niche_cluster_loadings_dataset_from_anadata
fig, axes = plot_niche_cluster_loadings_dataset_from_anadata(ana_data=ana_data)
# fig.savefig('figures/Spatial_niche_clustering_loadings.png', dpi=100) # uncomment this line if you want save the results
Niche Cluster Connectivity¶
from ONTraC.analysis.niche_cluster import plot_niche_cluster_connectivity_from_anadata
fig, axes = plot_niche_cluster_connectivity_from_anadata(ana_data=ana_data)
# fig.savefig('figures/Niche_cluster_connectivity.png', dpi=300) # uncomment this line if you want save the results
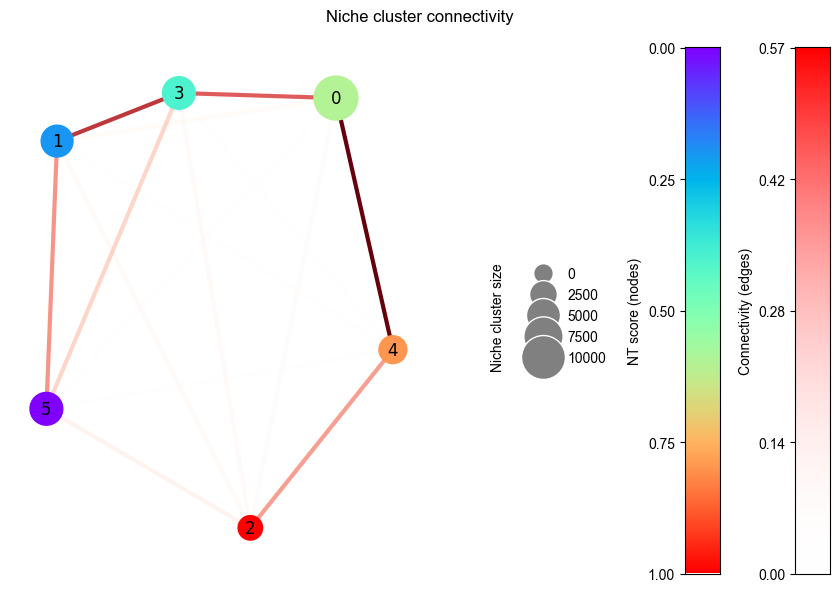
Note
This figure show the spatial connectivity between each niche cluster which will be used for niche trajectory construction.
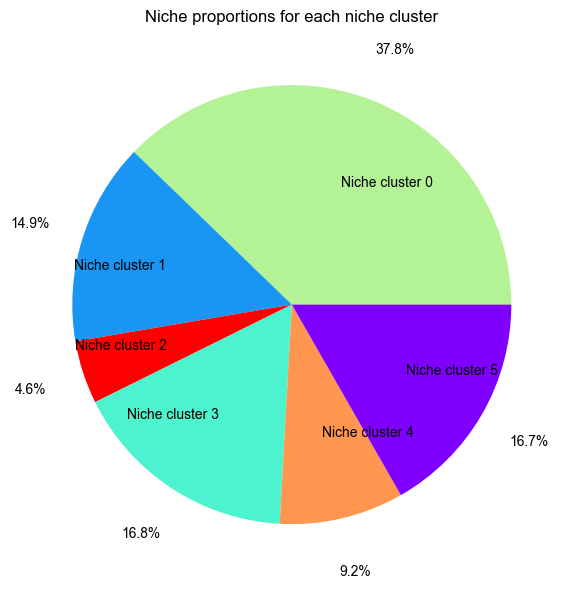
Niche Cluster Proportion¶
from ONTraC.analysis.niche_cluster import plot_cluster_proportion_from_anadata
fig, ax = plot_cluster_proportion_from_anadata(ana_data=ana_data)
# fig.savefig('figures/Niche_cluster_proportions.png', dpi=300) # uncomment this line if you want save the results
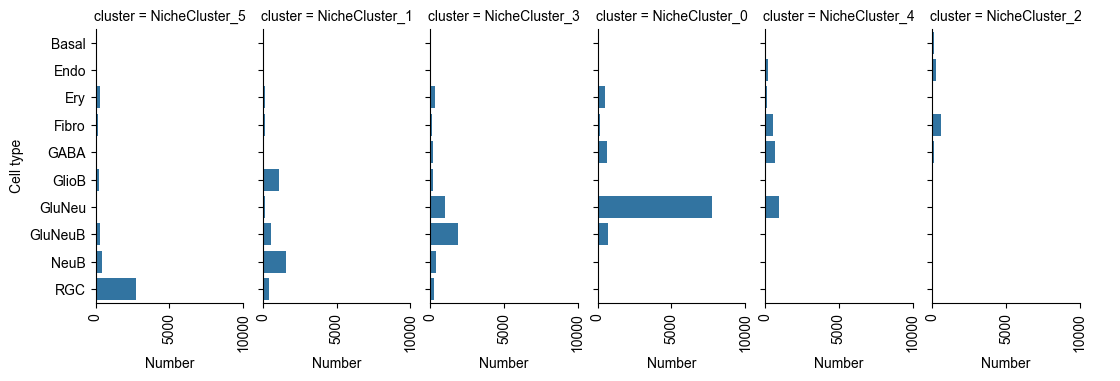
Cell Type X Niche Cluster¶
Number of Cells of Each Cell Type in Each Niche Cluster¶
from ONTraC.analysis.cell_type import plot_cell_type_loading_in_niche_clusters_from_anadata
g = plot_cell_type_loading_in_niche_clusters_from_anadata(ana_data=ana_data)
# g.savefig('figures/cell_type_loading_in_niche_clusters.png', dpi=300) # uncomment this line if you want save the results
Cell Type Composition Normalized by Each Niche Cluster¶
from ONTraC.analysis.cell_type import plot_cell_type_com_in_niche_clusters_from_anadata
fig, ax = plot_cell_type_com_in_niche_clusters_from_anadata(ana_data=ana_data)
# fig.savefig('figures/cell_type_composition_in_niche_clusters.png', dpi=300) # uncomment this line if you want save the results
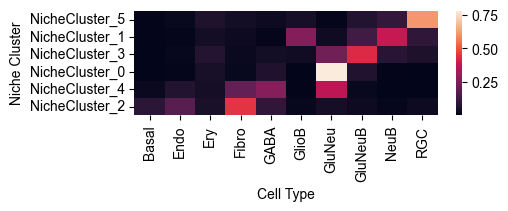
Note
This heatmap show the cell type composition within each niche cluster. Sum of each row equals to 1.
Cell Type Composition Normalized by Each Cell Type¶
from ONTraC.analysis.cell_type import plot_cell_type_dis_across_niche_cluster_from_anadata
fig, ax = plot_cell_type_dis_across_niche_cluster_from_anadata(ana_data=ana_data)
# fig.savefig('figures/cell_type_dis_across_niche_cluster.png', dpi=300) # uncomment this line if you want save the results
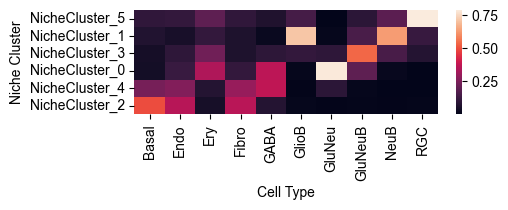
Note
This heatmap show the cell type distribution across niche clusters. Sum of each column equals to 1.
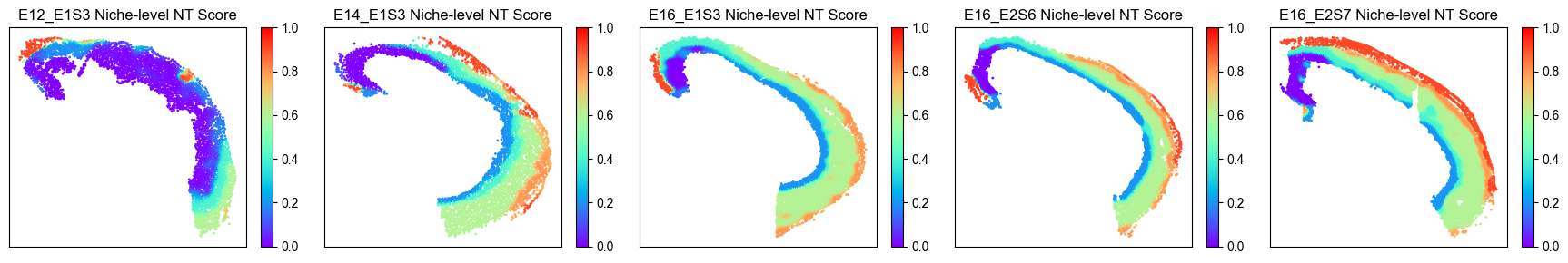
Spatial Niche-level NT Score Distribution¶
from ONTraC.analysis.spatial import plot_niche_NT_score_dataset_from_anadata
fig, ax = plot_niche_NT_score_dataset_from_anadata(ana_data=ana_data)
# fig.savefig('figures/niche_NT_score.png', dpi=200) # uncomment this line if you want save the results
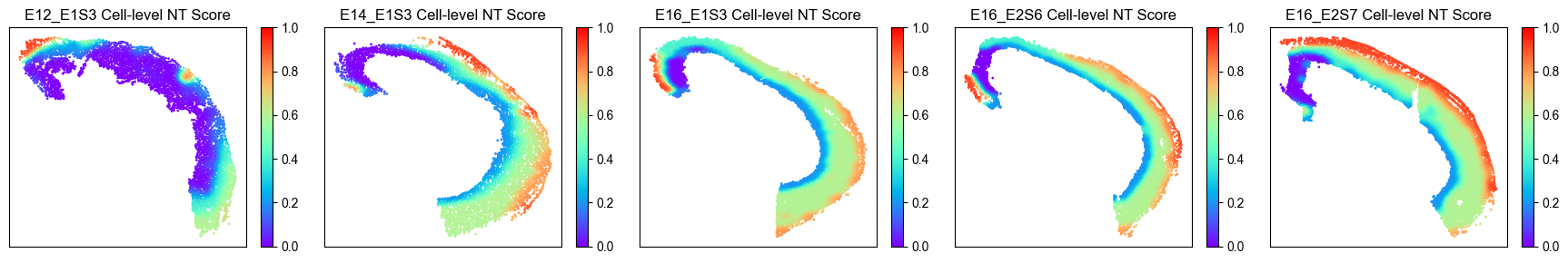
Spatial Cell-level NT Score Distribution¶
from ONTraC.analysis.spatial import plot_cell_NT_score_dataset_from_anadata
fig, ax = plot_cell_NT_score_dataset_from_anadata(ana_data=ana_data)
# fig.savefig('figures/cell_NT_score.png', dpi=200) # uncomment this line if you want save the results
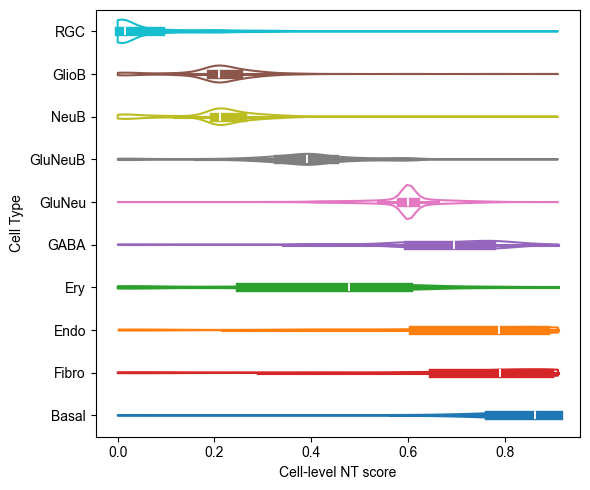
Cell-level NT Score Distribution for Each Cell Type¶
from ONTraC.analysis.cell_type import plot_violin_cell_type_along_NT_score_from_anadata
fig, ax = plot_violin_cell_type_along_NT_score_from_anadata(ana_data=ana_data,
order=['RGC', 'GlioB', 'NeuB', 'GluNeuB', 'GluNeu', 'GABA', 'Ery', 'Endo', 'Fibro', 'Basal'], # change based on your own dataset or remove this line
)
# fig.savefig('figures/cell_type_along_NT_score_violin.png', dpi=300) # uncomment this line if you want save the results